DNA拓扑异构酶是存在于细胞核内的一类内切酶,能够催化DNA链的断裂和结合,从而控制DNA的拓扑状态。研究发现,肿瘤细胞中拓扑异构酶呈现出不受其它因素影响的高表达现象,抑制其活性可阻止肿瘤细胞的增殖,因此成为抗肿瘤药物开发的重要靶点。目前,临床上使用的拓扑异构酶Ⅰ抑制剂喜书碱(Camptothecin)和拓扑异构酶Ⅱ抑制剂依托泊苷(Etoposide)和阿霉素(Doxorubicin)均因毒性和耐药性问题限制了其广泛应用。研究表明,同时抑制拓扑异构酶Ⅰ和Ⅱ有望解决该类抑制剂耐药问题。然而,目前并没有拓扑异构酶Ⅰ和Ⅱ双靶点抑制剂成功上市。因此,开发活性好、毒性低、对耐药细胞敏感的拓扑异构酶Ⅰ和Ⅱ双靶点抑制剂极具潜力。
Calothrixin A是从海洋蓝藻中分离提取的天然产物,能微弱地抑制拓扑异构酶Ⅰ和Ⅱ,但该分子溶解度差。重点实验室贺耘/张少林及其合作者通过合理药物设计策略,在Calothrixin A中引入亲水的氢键供体基团,不仅改善了Calothrixin A衍生物的溶解度,还增强了分子与靶点拓扑异构酶Ⅰ和Ⅱ的结合能力。体内外活性测试表明,衍生物F16比临床使用药物毒性更低,并且对阿霉素耐药细胞株敏感。
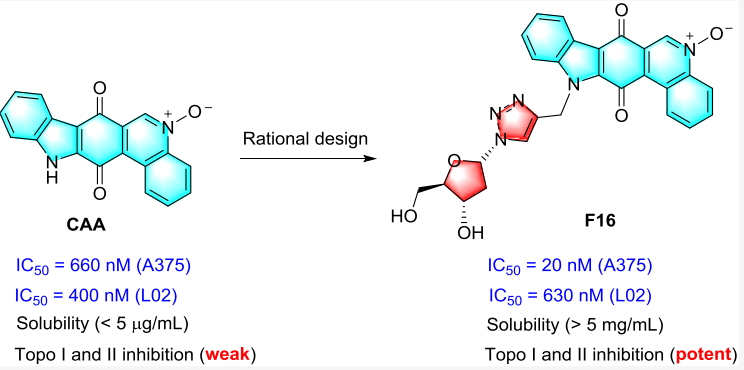
相关研究成果以“Optimization of the Natural Product Calothrixin A to Discover Novel Dual Topoisomerase I and II Inhibitors with Improved Anticancer Activity”为题,发表在药物化学领域权威杂志《Journal of Medicinal Chemistry》上。
文章链接:https://pubs.acs.org/doi/10.1021/acs.jmedchem.2c00615
(天然产物全合成与创新药物研究重庆市重点实验室)

 当前位置:
当前位置: